|
Allgemeines
Publikationen
|
Druck-Version
Neues aus der Forschung
Unsere haarigen Vettern
Neues von der genetischen Verwandtschaft zwischen Mensch und Schimpanse
Die Ermittlung von DNA-Sequenzen, eine Technologie, die 1965 von Frederick SANGER entwickelt wurde, war bis in die 1990er Jahre eine mühevolle Angelegenheit. Ende der 1980er Jahre fanden die ersten Genbanken (seinerzeit noch auf Disketten!) Verbreitung, wobei man erst viel später Vorstellungen über den Evolutionsprozess auf DNA-Sequenzebene zu entwickeln begann. Schnell wurde klar, dass sich DNA-Abschnitte und Sequenzpositionen, die funktional sind, langsamer ändern als funktionslose Bereiche ("Junk-DNA"): Je wichtiger bzw. zentraler eine Funktion ist, umso stärker ist der Selektionsdruck auf die kodierenden Sequenzen. Das entsprach den theoretischen Erwartungen und stellte keine Überraschung dar. Die Detailfragen blieben allerdings zunächst offen, weil schlichtweg die Daten fehlten, daher hatten wir keine Ahnung von der Dynamik komplexer Genome.
Die "Post-Genomics-Ära"
Etwa ab dem Jahre 2000 lag genügend genomische Sequenzinformation vor, um große Abschnitte der menschlichen Erbinformation zu analysieren. In den Jahren 2000-2010 kamen weitere Genome hinzu, die miteinander verglichen werden konnten. Ferner wurden immer größere Bereiche des Genoms verschiedener Menschen sequenziert, so dass auch verschiedenen Ethnien vergleichend analysiert werden konnten. Durch die Fortschritte der Molekularen Genetik wurden auch die molekularen Mechanismen der Vererbung und die Gründe für genetische Stabilität und Instabilität immer besser verstanden. Wie nicht anders zu erwarten, stellt die Evolution auf Genomebene ein äußerst komplexes und vielschichtiges Geschehen dar, das wir mittlerweile im Ansatz einigermaßen gut verstehen.
Repeats, hot spots, Mitochondriom, Y-Chromosom
In den Genomen der Säuger (und nicht nur dort) besteht ein mehr oder weniger großer Teil (bei Säugern gut die Hälfte!) (1) aus so genannten Repeats, also aus Sequenzanteilen, die mehrfach wiederholt vorkommen (s. u., Abb.1).Einige Repeats, wie die Zentromer- und Telomer-Sequenzen, sind von großer Wichtigkeit für die chromosomale Integrität: Telomere dienen dem Schutz der Chromosomenenden, und die Zentromere spielen eine wichtige Rolle bei der Verteilung der Chromosomen bei der Zellteilung (Mitose / Meiose). Mittels der Zentromere "klinken" sich sozusagen die Chromosomen in die Mitosespindel ein, so dass die Verteilung der Chromosomen auf die Tochterzellen geregelt und geordnet erfolgen kann. Einige der Repeats kodieren funktionale Produkte, z.B. liegen die Gene für die rRNA oft repetitiv vor. Die meisten Repeats sind allerdings nach heutigem Wissen funktionslos, sie werden oft als "Junk-DNA" ("Müll") bezeichnet. Bei dieser Beurteilung muss man allerdings vorsichtig sein: Die Evolution "weiß" nicht, dass diese Sequenzen ursprünglich funktionslos waren, und was in einem biologischen System vorhanden ist, unterliegt notwendigerweise und automatisch der Selektion. Daher nimmt es nicht Wunder, dass auch einige dieser Repeats vermutlich strukturelle Aufgaben besitzen (genauer: mittlerweile übernommen haben). Allerdings hängen diese Funktionen nicht so stark an einer definierten Sequenz, so sich dass solche Repeats relativ schnell ändern: Sie haben eine hohe Evolutionsgeschwindigkeit, was sich schon zwischen verschiedenen menschlichen Ethnien oder sogar Individuen bemerkbar macht.
Solche Repeats sind Bereiche mit sehr hoher Mutationsfrequenz; die sog. STR-Klasse (Abb.1) mutiert mit der Häufigkeit einiger Promille pro Generation (!), einige sind sogar derart instabil, dass sie kaum eine einzige Generation lang (!) unverändert bleiben. Die Mechanismen (helical slippage, unequal crossover, um nur einmal die Fachbegriffe zu nennen, die hier nicht im Detail erläutert werden können) sind bekannt.

TPOX-STR (GenBank Acc.Nr. M68651)
Eine Sorte von Repeats sind die sog. Tandem Repeats, das sind DNA-Elemente, die nicht im Genom verstreut sind, sondern sich als (identische oder ähnliche) Kopien unmittelbar aneinander anschließen. Die Wiederholungseinheiten sind kurz (wenige Nukleotide oder sogar nur ein einziges) oder länger: Einheiten von Hunderten von Basen kommen vor. Kurze Einheiten - so wie das hier abgebildete GAAT-Repeat - bezeichnet man als short tandem Repeats (kurz: STRs). Da solche STRs bereits in der menschlichen Bevölkerung z. T. erheblich variieren, werden sie für Abstammungsgutachten und forensische Analysen benutzt (die Pfeile in der Abbildung bezeichnen Primer-Bindestellen für forensische PCR-Analysen, was hier nicht im Detail ausgeführt werden kann).
Abb.1: Beispiel für einen menschlichen STR-Lokus.
Darüber hinaus gibt es weitere Bereiche und Stellen im Genom, die sich ebenfalls schneller ändern als der Durchschnitt, ohne dass die Gründe hierfür bekannt wären; hier sind also noch spannende Forschungsergebnisse zu erwarten.
"Mitochondriom" (oft abgekürzt mit mtDNA) nennt man die Erbinformation der Mitochondrien. Diese Zellorganellen sind die "Kraftwerke" der Zellen, sie produzieren den Löwenanteil der zellulären Energie (in Form von ATP) hauptsächlich durch Verbrennung von Lipiden und Kohlenhydraten (genau genommen von Reduktionsäquivalenten, das sind sozusagen "gebundene, transportable Wasserstoffatome"). Mitochondrien - darauf deutet eine Vielzahl voneinander unabhängiger Fakten hin - sind aus ehemals frei lebenden Bakterien hervorgegangen, die vor gut einer Milliarde Jahren eine Symbiose mit einer Ur-Eukaryontenzelle eingingen. Das genetische System der Mitochondrien ist im Vergleich zum Zellkern simpler und weniger leistungsstark; die Mitochondrien rekombinieren (fast) nie und werden bei höheren Säugern (fast) immer über die Mutter vererbt. Aus diesen Gründen evolviert die mtDNA sehr viel schneller als das Genom des Zellkerns - so schnell, dass man anhand der Unterschiede in der mtDNA verschiedener Ethnien die Wanderungsbewegungen der frühen Menschheit rekonstruieren konnte (Stichwort "mitochondriale Eva"). Wie nicht anders zu erwarten ist die menschliche mtDNA wegen der hohen Mutationsraten von der Schimpansen-mtDNA deutlich verschiedener als es die Genome des Zellkerns sind.
Auch das Y-Chromosom - dasjenige Geschlechtschromosom, welches "den Mann zum Manne" macht - unterliegt einem besonderen Vererbungsmodus: Während Frauen mit XX über zwei gleiche Geschlechtschromosomen verfügen, tragen Männer die Kombination XY, wobei beide Chromosomen einander ziemlich unähnlich sind (Y ist auch wesentlich kleiner). Das bedeutet ferner, dass das Chromosom stets vom Vater an die Söhne vererbt wird und dass Y-Chromosomen nicht rekombinieren können, weil sie (fast) immer alleine auftreten.
Dies hat erhebliche Auswirkungen auf die evolutive Entwicklung: Y-Chromosomen (das der Säuger entstand vor einigen 100 Mio. Jahren durch Modifikation eines X-Autosoms) verlieren Zug um Zug die meisten ihrer Gene und erfahren dann am Ende auf dem größten Teil ihrer Sequenz keinen Selektionsdruck mehr (vgl. CHARLESWORTH/CHARLESWORTH 2000; eine Übersicht findet sich unter de.wikipedia.org/wiki/Y-Chromosom). (2) Hinzu kommt, dass Männer wegen der lebenslang anhaltenden Spermienbildung etwa doppelt so viele Mutationen an ihre Nachkommen vererben wie Frauen. Deshalb und weil das Y-Chromosom allein in der Linie männlicher Nachkommen vererbt wird, evolviert es deutlich schneller. Beides sind Gründe dafür, dass sich auf Y-Chromosomen repetitive Sequenzen in oftmals wesentlich höherer Dichte als auf den Nicht-Geschlechtschromosomen (den Autosomen) ansammeln. Daher sind auch bereits innerhalb der menschlichen Bevölkerung die Unterschiede zwischen den Y-Chromosomen größer als zwischen den Autosomen. In der Tat zeigten KURODA-KAWAGUCHI et al. (2001), dass bei unfruchtbaren Männern eine bestimmte Art von Deletion auf dem Y-Chromosom oftmals und unabhängig voneinander vorkommt: Ein klarer Hinweis (mittlerweile einer von vielen) auf die vergleichsweise hohe Instabilität des Y-Chromosoms.
Weitere Daten kamen hinzu, als SKALETSKY et al. (2003) eine detaillierte Studie des menschlichen Y-Chromosoms vorlegten: Sie zeigten, dass - den Erwartungen durchaus entsprechend - das menschliche Y eine sehr hohe Repeat-Dichte aufweist und innerhalb der letzten 4 Mio. Jahre durch Translokation X-Sequenzen inkorporiert hat, dass also im Laufe der menschlichen Evolution Bereiche des X-Geschlechtschromosoms auf Y kopiert wurden.
Das Y-Chromosom von Mensch und Schimpanse im Vergleich
Weil Y-Chromosomen gerade wegen der hohen Repeat-Dichte technisch schwer zu sequenzieren sind (genauer: die gewonnene Sequenzdaten sind schwerer zu assemblieren, d.h. "zusammenzupuzzeln", weil große Bereiche in vielfacher Wiederholung, als Repeats, vorliegen), lagen bisher außer vom Menschen nur wenig Y-Sequenzdaten von anderen Säugern vor. Zwar ist durch Analyse des menschlichen Genoms z.B. erkennbar, welche Chromosomen-Anteile (und damit welche Gene) von X auf Y übertragen wurden (s. o.), aber es war unklar, ob die Evolution des menschlichen Y vielleicht besonders "stürmisch" oder trotz der massiven Umbauten doch noch im Relation zu anderen Säugern vergleichsweise ruhig verlaufen ist: Die extrem hohe Dichte an Repeats ist mit Sicherheit ganz wesentlich für die evolutionäre Instabilität verantwortlich - aber in welchem Ausmaß, das war noch die Frage (vgl. Abb.2).
Eine erste Antwort lieferte vor kurzem eine Arbeit von HUGHES et al. (2010), in der fast die komplette Sequenz des Schimpansen-Y-Chromosoms vorgelegt und ein umfassender Vergleich durchgeführt wurde. Der prinzipielle Aufbau des Y-Chromosoms der Schimpansen entspricht wie erwartet dem des Menschen, auch die vertretenen Sequenzklassen sind dieselben (allerdings sind manche Familien auf Mensch oder Schimpanse beschränkt). Ferner lassen sich auf dem menschlichen Y-Chromosom Bereiche nachweisen, die (beurteilt nach ihrer Sequenzähnlichkeit) innerhalb der letzten 4 Millionen Jahren vom (vor-) menschlichen X-Chromosom auf das Y-Chromosom übertragen wurde - also zu einer Zeit, als die menschliche Abstammungslinie schon von der Schimpansenlinie getrennt hat. Diese Bereiche treten erwartungsgemäß auf dem Schimpansen-Y nicht auf. Ebenfalls zu vermuten war, dass insbesondere die repetitiven Abschnitte (Repeats, "ampliconic sequences") schneller evolvieren als die nicht-repetitiven. Sequenz-Zuwachs und -Verlust sowie Umstrukturierungen durch Translokation und Inversion (3) sind in großem Ausmaß über das gesamte Y-Chromosom zu beobachten. Auch dies war nicht unvermutet, haben doch schon REPPING et al. (2006) gezeigt, dass das menschliche Y-Chromosom innerhalb der letzen 100.000 Jahre in verschiedenen Ethnien ganz erhebliche Änderungen erfahren hat (eben solche Transpositionen, Translokationen und Inversionen). Es war dann für HUGHES et al. aber nochmals beeindruckend zu sehen, wie instabil die Y-Chromosomen im Vergleich zwischen Mensch und Schimpanse tatsächlich sind. Diese Ergebnisse werden zu einer Weiterentwicklung der Theorie der Geschlechtschromosomen-Evoluton führen. In Zukunft werden sicherlich weitere Säuger-Y-Chromosomen komplett sequenziert und verglichen werden; insbesondere die Rolle repetitiver Sequenzen für chromosomale Umstrukturierungsprozesse wird man dann besser verstehen und beschreiben können (Abb.2). HUGHES et al. haben erste Einblicke in dafür verantwortlichen die molekularen Mechanismen gewonnen, was den Rahmen einer News allerdings sprengen würde.
Abb.2: Beispiel für Genverlust durch "unequal crossover". Quelle: ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=hmg&part=A2004
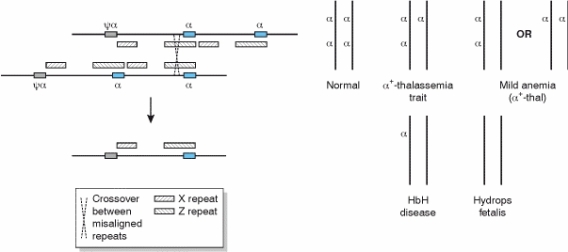
Die Geschichte Y-chromosomaler Veränderungen ist zu komplex, um sie in Kürze darzustellen, daher hier ein Beispiel von einem Autosom: "Thalassämien" sind Erbkrankheiten, die auf Genverlust von HämoglobinGenen beruhen. Das menschliche Chromosom 16 trägt zwei intakte Kopien sowie eine defekte Kopie Psi der Alpha-Kette. Größere Bereiche sind einander hochgradig ähnlich (graue Balken), so dass bei der Keimzellbildung (Meiose) zu einem fehlerhaften Austausch zwischen den elterlichen Chromosomen kommen kann, zum ungleichen Stückaustausch ("unequal crossover"). Dadurch gehen chromosomale Bereiche verloren oder werden verdoppelt (s. Abbildung); wenn die repetitiven Bereiche auf verschiedenen Chromosomen liegen, kann es zu Stückaustauschen zwischen verschiedenen Chromosomen kommen (WEATHERALL et al. 1995). Derartige Mutationen auf Autosomen, also auf den "normalen Chromosomen", haben fast immer mehr oder weniger große Auswirkungen. Da das Y-Chromosom weitgehend "genleer" ist und einen sehr hohen Repeat-Anteil aufweist, kommen solche Umstrukturierungen auf Y sehr viel häufiger vor, werden dabei aber nur selten bemerkt, weil sie sich meist nicht auswirken.
... Und was macht Wort und Wissen daraus?
Unter dem Titel "Neue Daten zum Y-Chromosom des Schimpansen" hat Wort und Wissen (W+W) eine News verfasst (BINDER 2010), in der es heißt:
"Nach gängigen Vorstellungen haben sich die Entwicklungslinien von Mensch und Schimpansen von einem hypothetischen gemeinsamen Vorfahren vor ca. 6 Millionen Jahren getrennt. Bisherige Genomvergleiche mit geringen Unterschieden (ca. 2 %) scheinen dies zu bestätigen. Die jetzt bei Y-Chromosomen dokumentierten Unterschiede entsprechen dagegen einer unabhängigen Entwicklung seit ca. 310 Millionen Jahren."
Das ist, so wie es dasteht, völlig falsch. Erstens evolviert aus oben genannten Gründen sowohl die mtDNA als auch das Y-Chromosom schneller als der Rest des Genoms, was sich bereits innerhalb der menschlichen Population zeigen lässt. Also sind erheblich größere Unterschiede in beiden Fällen zu erwarten und nicht, wie BINDER suggeriert, überraschend und unverständlich. Zweitens würden, wenn man tatsächlich so rechnete, die Evolutionsraten vollkommen unterschiedlicher Mutationstypen in einen Topf geworfen. Drittens hat W+W die Zeile in der Publikation in grob irreführender Weise übersetzt. Wörtlich steht in der Publikation das Folgende:
"we found that the degree of similarity between orthologous chimpanzee and human MSY sequences (98.3% nucleotide identity) differs only modestly from that reported when comparing the rest of the chimpanzee and human genomes (98.8%) (...) Indeed, at 6 million years of separation, the difference in MSY gene content in chimpanzee and human is more comparable to the difference in autosomal gene content in chicken and human, at 310million years of separation"
Die korrekte Übertragung ins Deutsche im Kontext und vor dem Hintergrund der in der Publikation vorgelegten Ergebnisse müsste demnach lauten: "Nach 6 Mio. Jahren der Trennung zwischen Mensch und Schimpanse haben die besonderen genetischen Verhältnisse in den Y-Chromosomen für Unterschiede in Struktur und Geninhalt gesorgt, wie sie bei Wirbeltieren in genomischer DNA ansonsten erst nach gut 300 Mio. Jahren auftreten. Dabei liegt der Grad der Sequenzdivergenz alignierbarer single-copy Gene wie erwartet in derselben Größenordnung wie bei Autosomen." (4)
BINDER weiter:
"Die heute bekannten DNA-Sequenzen von Mensch und Schimpansen verlangen eine sehr differenzierte Erklärung und unterstützen nicht einfach ein etabliertes Bild von der gemeinsamen Abstammung von einem gemeinsamen Vorfahren. (…) Die mittlerweile ermittelten Daten von Y-Chromosomen sperren sich somit gegen eine Vereinnahmung für eine klassische Vorstellung der Abstammung von Mensch und Schimpanse von einem hypothetischen gemeinsamen Vorfahren. Die gravierenden Unterschiede in der Struktur des Y-Chromosoms werfen neue Fragen nach deren Ursprung auf; die bekannten und üblicherweise angenommenen Mechanismen würden eine deutlichere Ähnlichkeit erwarten lassen."
Selbst wenn manche Autoren eine geringere Abweichung der Y-Chromosomen erwartet haben mögen, so ist doch diese Aussage frei erfunden und dem Text nirgends zu entnehmen, im Gegenteil. Bedauerlicherweise ist dies (wieder einmal) ein Beispiel für die kreationistische Technik, einen wissenschaftlichen Text "zu lesen wie der Teufel die Bibel" (skandinavisches Sprichwort) und, wenn das noch nicht reicht, Dinge hineinzulesen, die gar nicht drin stehen. Falls Wort und Wissen von der Fachwelt ernst genommen werden möchte, sollte die Vereinigung künftig besser auf diese Technik verzichten.
Fazit
Evolution ist ein vielschichtiges und extrem komplexes Geschehen. Selbst wenn nur eine einzige Ebene (hier: die genetische) betrachtet wird, sind die Vorgänge ungeheuer kompliziert.
Verschiedenen Mutationsarten wie Mikromutationen (Basenaustausche, kleine Deletionen oder Insertionen) und Mutationen auf chromosomaler Ebene (Translokation, Deletion, Insertion größerer Bereiche etc.) finden mit unterschiedlicher Häufigkeit statt. Hinzu kommt, dass aufgrund unterschiedlicher Mutationsmechanismen manche Sequenzklassen (methylierte CG-Dinukleotide, Repeats) sehr viel schneller mutieren als andere. Ferner ist der Modus der Vererbung ein wichtiger Punkt, der über das evolutionäre Schicksal entscheidet: mitochondriale DNA (mtDNA) und das Y-Chromosom können bei der sexuellen Fortpflanzung (meiotische Bildung der Keimzellen) nicht rekombinieren, was u. a. auf die Häufigkeit von Genverlusten beeinflusst. Hinzu kommt bei der mtDNA ein ungenauer arbeitendes DNA-Reparatursystem. Schließlich sind die Gendichte sowie der Selektionsdruck Faktoren, die das evolutive Schicksal eines Chromosoms mit bestimmen: je geringer der Einfluss beider Faktoren, desto weniger fallen chromosomale Mutationen ins Gewicht und umso stärker wird die chromosomale Struktur evolutiv variieren.
Im Rahmen einer Untersuchung von HUGHES et al (2010) wurden die Y-Chromosomen von Mensch und Schimpanse miteinander verglichen. Es zeigt sich aus den oben angeführten Gründen, dass die genetischen Unterschiede sehr viel größer sind als im Rest der Kerngenome. Wie nicht anders zu erwarten, zeigen hingegen Y-chromosomale Gene, die wichtige Funktionen (Spermienreifung, Geschlechtsbestimmung) erfüllen trotz der beträchtlichen Unterschiede der chromosomalen Y-Strukturen denselben hohen Ähnlichkeitsgrad wie die "normalen" Chromosomen, die Autosomen.
Die kreationistische Studiengemeinschaft Wort und Wissen behauptet, dass die großen strukturellen Unterschiede der Y-Chromosomen die enge Verwandtschaft von Mensch und Schimpanse wieder in Frage stelle. Dieser Schluss kann allerdings nur unter Missachtung der genetischen Grundlagen und Zusammenhänge gezogen werden und vor allem dadurch, dass in die Veröffentlichung von HUGHES et al (2010) Aussagen hinein gelesen werden, die in dieser Arbeit definitiv nicht drin stehen.
Literatur
BINDER, H. (2010) Wie ähnlich sind Mensch und Schimpanse? Neue Daten zum Y-Chromosom des Schimpansen. News der kreationistischen Plattform Genesisnet, www.genesisnet.info/index.php?News=147.
CHARLESWORTH, B.; CHARLESWORTH. D. (2000) The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 355(1403), 1563-1572. www.ncbi.nlm.nih.gov/pmc/articles/PMC1692900/?tool=pubmed; www.ncbi.nlm.nih.gov/pmc/articles/PMC1692900/pdf/11127901.pdf
HUGHES, J.F.; SKALETSKY, H.; PYNTIKOVA, T. et al. (2010) Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463(7280), 536-539.
KURODA-KAWAGUCHI, T.; SKALETSKY, H. et al. (2001) The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nature Genet. 29, 279-286.
REPPING, S.; VAN DAALEN, S.K.; BROWN, L.G. et al. (2006) High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet. 38(4), 463-467.
SKALETSKY, H.; KURODA-KAWAGUCHI, T. et al. (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825-837.
Weitere Literatur zum Thema
CHEUNG, J.; WILSON, M.D.; ZHANG, J.; KHAJA et al. (2003) Recent segmental and gene duplications in the mouse genome. Genome Biol. 4(8), R47. www.ncbi.nlm.nih.gov/pmc/articles/PMC193640.
HASTINGS, P.J.; LUPSKI, J.R.; ROSENBERG, S.M.; IRA, G. (2009) Mechanisms of change in gene copy number. Nature Reviews Genetics 10, 551-564.
PARNIEWSKI, P.; STACZEK, P. (2002) Molecular Mechanisms of TRS Instability. www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=eurekah&part=A13297
Abschnitt des Internet-Lehrbuchs "Gene Expression", www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=eurekah&part=part31.xml.
STRACHAN, T.; READ, A.P. (1999, Hg.) Human Molecukar Genetics 2. Garland Science. Internet-Version: www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=hmg.
WOLFE, J. (2006) Sex Determination. Internet-Lecture, Biology of Development course BiolB250, London's global university. www.ucl.ac.uk/~ucbhjow/b250/sex_determination.html.____________________________________________
Fußnoten
(1) Die genaue Zahl ist schwer zu ermitteln. Insbesondere bei Repeats, die über das Genom verstreut sind, gilt: je mehr Zeit vergangen ist seit die betreffenden Kopien auseinander hervorgegangen sind, desto unähnlicher sind sie sich geworden. Daher wird man bei weitem nicht alle repetitiven Sequenzen als solche erkennen können, was hier nicht weiter vertieft werden kann.
(2) Inzwischen kennen wir etliche Beispiele für ältere, aber auch für erst wenige Millionen Jahre alte Y-Chromosomen in verschiedenen biologischen Arten. All diese Y-Chromosomen neigen grundsätzlich zum Genverlust und zur Degeneration. Diese Arbeit bietet einen Überblick über den Stand der Forschung und die Modelle, welche zur Beschreibung und Erklärung des Genverlusts mittlerweile erarbeitet sind. [Übersichtsartikel].
(3) Translokation: verschieben, Inversion: verdrehen, Deletion: entfernen, Duplikation: verdoppeln kleinerer oder größerer Abschnitte eines Chromosoms.
(4) Konkret: Punktmutationen (Basenaustausche) geschehen häufiger als kleinere Deletionen oder Insertionen, und diese wiederum weitaus häufiger als große chromosomale Umstellungen (Translokationen oder Inversionen großer Abschnitte). Wenn man solche Mutationsraten zur Bestimmung verwandtschaftlicher Verhältnisse heran zieht, muss man selbstverständlich alle Typen separat betrachten und die Mutationsraten auch separat eichen. Das ist z.B. möglich, indem man die unterschiedlichen Raten innerhalb der heutigen menschlichen Bevölkerung bestimmt und dann - mit der gebotenen Vorsicht - extrapoliert. Das Vermengen dieser verschiedenen Raten im Rahmen einer bioinformatisch-statistischen Analyse ist eine mathematische Todsünde.
Autor: Andreas Beyer

© AG Evolutionsbiologie des VdBiol. 11.05.2010